2026年2月,国家血液系统疾病临床医学研究中心、苏州大学附属第一医院、苏州大学造血干细胞移植研究所的吴德沛教授、徐杨教授和储剑虹教授团队在《Advanced Science》(IF=14.1)杂志在线发表了题为 “Targeting PRKCN, an essential driver orchestrating mTOR-IRF4 axis independently of kinase activity, in multiple myeloma”的研究论文。该研究首次系统揭示了PRKCN在多发性骨髓瘤中的关键致癌作用及其不依赖于激酶活性的独特调控机制,为临床治疗提供了新的潜在靶点和候选药物。

尽管新药不断涌现,但多发性骨髓瘤的耐药和复发仍是临床治疗的巨大挑战。因此,深入理解其发病机制,寻找驱动疾病进展的核心分子,对于开发更有效的疗法至关重要。
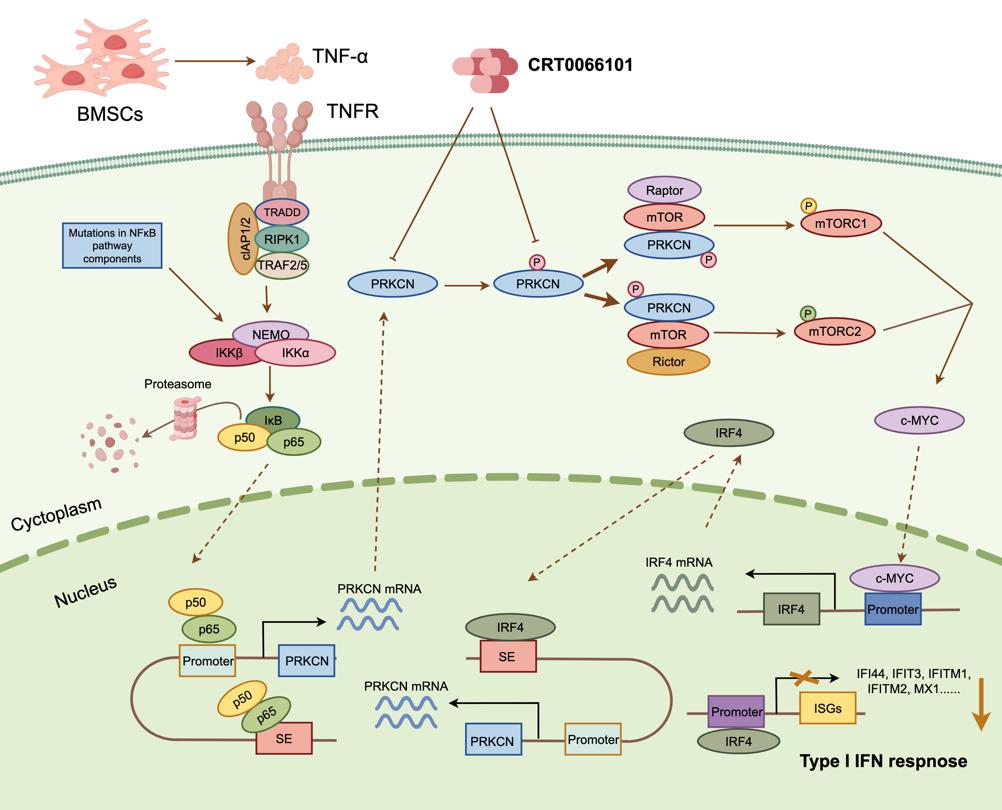
PRKCN(又称PKD3)属于蛋白激酶D家族,在实体瘤中被报道参与肿瘤进展,但其在血液肿瘤,特别是多发性骨髓瘤中的角色,此前一直是个谜。
核心发现:揭示PRKCN在骨髓瘤中的关键作用
本研究通过一系列精密的分子生物学和细胞生物学实验,结合动物模型和临床样本分析,清晰地描绘了PRKCN在多发性骨髓瘤中的“画像”:
1. PRKCN是骨髓瘤的“坏分子”
研究发现,PRKCN是一个由超级增强子(SE) 驱动的基因,在多发性骨髓瘤细胞和患者样本中异常高表达,且与患者的不良预后密切相关。高表达PRKCN的患者,其无事件生存期(EFS)、总生存期(OS)和无进展生存期(PFS)均显著缩短。
2.独特的调控机制:NF-κB信号通路的关键下游
骨髓微环境中的炎症因子(如TNF-α)或肿瘤细胞内在的NF-κB信号通路激活,可直接与PRKCN的启动子和超级增强子结合,“命令”癌细胞大量生产PRKCN。这表明PRKCN是NF-κB这一经典致癌通路的关键效应分子。
3.核心功能:驾驭mTOR-IRF4“致癌轴”
功能实验证实,PRKCN能强力促进骨髓瘤细胞的增殖、成瘤能力和耐药性,同时抑制细胞凋亡。其核心机制在于,PRKCN通过与mTOR蛋白直接结合,激活了mTORC1和mTORC2两条关键信号通路,进而上调了IRF4——一个公认的骨髓瘤“总开关”和生存必需因子的表达。
4.颠覆性发现:不依赖激酶活性的新“职场路径”
这可能是本研究最令人惊讶的发现!通常,蛋白激酶通过其激酶活性“磷酸化”下游蛋白来传递信号。但本研究发现,PRKCN激活mTOR-IRF4轴并促进细胞生长的功能,竟然完全不依赖其固有的激酶活性!相反,它依赖于其活化环(activation loop)的磷酸化状态。这意味着,PRKCN更像是一个“脚手架”蛋白,通过其磷酸化的活化环来招募和激活下游信号复合物。这一发现颠覆了传统认知,为靶向PRKCN提供了全新的思路:与其抑制其激酶活性,不如破坏其活化环磷酸化。
5.形成一个自我强化的“致癌环路”
研究还发现,PRKCN上调IRF4,而IRF4反过来又能结合到PRKCN的超级增强子上,直接促进PRKCN的转录。这就形成了一个PRKCN↔IRF4的正反馈调节环路,像滚雪球一样不断放大致癌信号。
6.潜在治疗策略:老药新用,效果显著
基于上述发现,研究团队测试了一种口服有效的泛PKD抑制剂——CRT0066101。结果显示,该药物在体外能有效抑制骨髓瘤细胞(包括对标准药物硼替佐米和来那度胺耐药的细胞)生长,并与标准药物产生协同效应。更重要的是,在多种动物模型(包括细胞系来源的异种移植模型和患者来源的异种移植模型)中,CRT0066101均展现出强大的抗肿瘤效果,且对正常细胞毒性较低。其作用机制正是破坏了PRKCN的活化环磷酸化,从而阻断了mTOR-IRF4致癌轴。
研究意义:为攻克难治性骨髓瘤指明新方向
本研究具有重要的科学意义和临床转化潜力:
发现新靶点:首次将PRKCN确立为多发性骨髓瘤的一个关键驱动基因和潜在治疗靶点。
提出新机制:揭示了PRKCN不依赖激酶活性、而依赖活化环磷酸化的独特作用模式,拓宽了对蛋白激酶功能多样性的理解。
建立新连接:阐明了从NF-κB到PRKCN,再到mTOR-IRF4的完整信号链条,加深了对骨髓瘤发病网络的认识。
提供候选药:证明了现有抑制剂CRT0066101在多发性骨髓瘤模型中的强大疗效,为其向临床转化提供了坚实的临床前证据。研究强调,未来或许可以开发专门靶向PRKCN活化环的抑制剂,以期获得更精准、更安全的疗效。
该研究得到国家重点研发计划、国家自然科学基金委等多个科研资金的支持。苏州大学的汤寇寇、蒋东鹏、柯鹏、卞小森为本文共同第一作者,吴德沛教授、徐杨教授、傅琤琤教授、储剑虹教授为本文共同通讯作者。
原文摘要:
Multiple myeloma (MM) remains incurable, necessitating development of novel therapeutic targets. Deregulated PRKCN is implicated in solid tumors, while its role in MM remains elusive. Here, PRKCN is identified as a super-enhancer-driven gene associated with adverse prognosis in MM. PRKCN is transactivated by NF-κB signaling intrinsically existing or exogenously provoked. Constitutive or inducible knockdown of PRKCN significantly impairs cell growth and tumorigenicity, while overcoming drug resistance. PRKCN harnesses IRF4 to exert its effect, and in turn, IRF4 directly induces PRKCN transcription, establishing a feed-forward IRF4-PRKCN circuit. Furthermore, PRKCN fosters IRF4 expression by activating mTORC1/C2 signaling pathways via physical interaction with mTOR. Surprisingly, PRKCN modulates mTOR-IRF4 axis and cell growth independently of its acknowledged kinase activity yet requiring activation loop phosphorylation. Intriguingly, PRKCN silencing evokes interferon signaling and confers increased sensitivity to interferon. Finally, targeting PRKCN with an orally bioavailable inhibitor suppresses MM cell growth and overcomes drug resistance in vitro, and elicits robust efficacy in cell line-derived xenografts and a patient-derived xenograft, which is connected with the mitigated PRKCN expression and activation loop phosphorylation as well as blunted mTOR-IRF4 axis. Collectively, our study delineates PRKCN function that links aberrant NF-κB signaling and mTOR-IRF4 axis, supporting clinically targeting PRKCN in MM.
原文链接:https://doi.org/10.1002/advs.202518975

